-
組蛋白乳糖化誘導的早衰在1-硝基并芘誘導的慢性阻塞性肺疾病中的作用
發布時間: 2025-12-05 點擊次數: 364次各位讀者好,今天為大家帶來一篇使用整合乳酸化+細胞衰老+糖酵解三大熱點并結合動物、細胞和分子層面實驗來驗證1-硝基芘(1-NP)暴露致慢性阻塞性肺疾病(COPD)的機制的潛在靶點和機制的高分文章,是由安徽醫大二院團隊2025年5月在Redox Biology發表的,題為“Histone lactylation-induced premature senescence contributes to 1-nitropyrene-Induced chronic obstructive pulmonary disease"。深入探究了1-硝基芘(1-NP)暴露致慢性阻塞性肺疾病(COPD)的潛在機制,闡明1-NP通過H3K14la-p53通路誘導肺上皮細胞衰老致COPD,為抑制乳酸生成提出治療策略。

發表雜志:
《Redox Biology》是氧化還原生物學領域的國際期刊,創刊于2013年,聚焦氧化還原信號調控、氧化應激與疾病機制等核心方向,是生命科學(尤其是生物化學、分子生物學、病理生理學及藥物研發領域)科研人員的重要學術交流平臺。

2025 年影響因子:11.9
ISSN:2213-2317
中科院分區:大類:生物(1 區 Top);小類:生物化學與分子生物學(1 區)、細胞生物學(1 區)
發文量:每年出版文章數平均約373篇
發表成本:APC為3,900美元(約2.8萬人民幣),作為中科院1區TOP期刊,性價比相對合理
審稿周期:該期刊以高效審稿著稱,從投稿到接收平均僅需37 天,大多數作者反饋審稿周期在1-3 個月范圍,遠快于同類高影響因子期刊
《Redox Biology》作為氧化還原生物學領域的 Top 期刊,其高影響因子、嚴格的審稿標準、廣泛的學科覆蓋 使其成為該領域原創研究和綜述的重要發表平臺。對于從事以下研究的科研人員,尤其推薦投稿:
1.氧化應激與疾病(癌癥、神經退行性疾病、心血管疾病等)的機制研究;
2.抗氧化藥物 / 制劑的細胞 / 動物實驗驗證;
3.redox 信號通路、線粒體 redox 代謝的分子機制探究;
4.氧化還原相關檢測技術或組學分析方法的創新。
研究背景:
1-硝基芘(1-NP)是常見環境污染物,具遺傳毒性和致癌性,可引發呼吸系統疾病。慢性阻塞性肺疾病(COPD)是第三大致死病因,傳統認為吸煙是主因,但環境污染物作用漸受關注。前期研究顯示1-NP可誘導肺泡細胞衰老,而細胞衰老與COPD密切相關。組蛋白乳酸化作為新型表觀遺傳修飾,調控基因轉錄和細胞衰老。然而1-NP與COPD的關聯及組蛋白乳酸化在其中的作用尚不明確,故本研究探討1-NP通過組蛋白乳酸化誘導肺上皮細胞衰老致COPD的機制。
本文研究1-硝基芘(1-NP)暴露致慢性阻塞性肺疾病(COPD)的機制,發現慢性1-NP暴露通過促進肺上皮細胞糖酵解和乳酸脫氫酶A(LDHA)表達,升高乳酸水平,誘導組蛋白H3K14乳酸化(H3K14la),進而激活p53轉錄,導致細胞周期停滯和早衰,最終引發COPD樣表型。抑制乳酸生成可緩解該過程,為COPD治療提供新靶點。
研究框架:
1.提出問題:
基于1-NP的肺毒性及組蛋白乳酸化調控基因轉錄的特性,提出“1-NP是否通過組蛋白乳酸化誘導肺上皮細胞衰老,進而導致COPD"的假設。
2. 研究框架:
從整體動物、細胞和分子層面,依次驗證1-NP致COPD樣表型、誘導肺上皮細胞衰老、激活糖酵解-乳酸-組蛋白乳酸化通路、H3K14la調控p53轉錄及抑制乳酸生成的干預效果。
3. 研究方法:
動物實驗采用C57BL/6小鼠動態吸入1-NP構建COPD模型,檢測肺功能和病理變化;細胞實驗用MLE-12細胞,結合siRNA干擾(LDHA、p53)、Western blot、RT-qPCR、SA-β-gal染色、流式細胞術等;分子機制通過CUT&Tag和ChIP-qPCR驗證H3K14la與p53啟動子結合。
4. 分析數據:
采用GraphPad Prism進行統計學分析,組間比較用t檢驗或ANOVA,相關性分析用Pearson檢驗,P<0.05為差異顯著。
5. 研究結論:
整合動物、細胞和分子實驗結果,闡明1-NP通過H3K14la-p53通路誘導肺上皮細胞衰老致COPD的機制,提出抑制乳酸生成的治療策略。
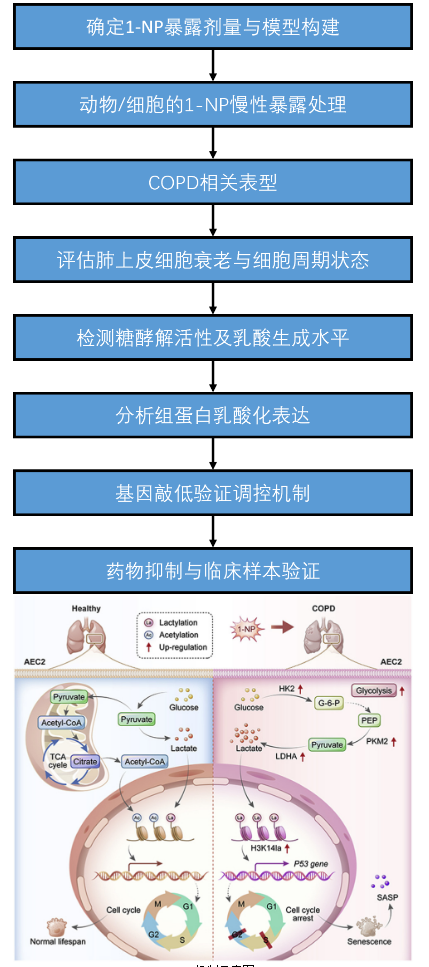
Fig.機制示意圖
結果解析:
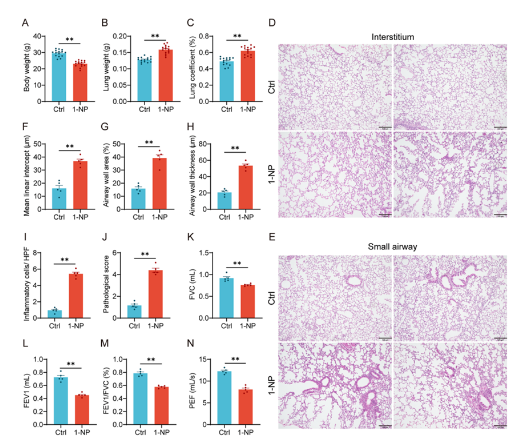
1. 1-NP暴露導致小鼠出現COPD樣表型
通過動態吸入暴露裝置讓小鼠長期接觸1-NP氣溶膠,結果顯示小鼠體重下降,肺重量及肺系數增加,肺泡結構受損、炎癥細胞浸潤,平均線性截距、氣道壁面積和厚度增大,肺功能指標(FVC、FEV1、FEV1/FVC、PEF)顯著降低,呈現出COPD樣病理和功能改變。
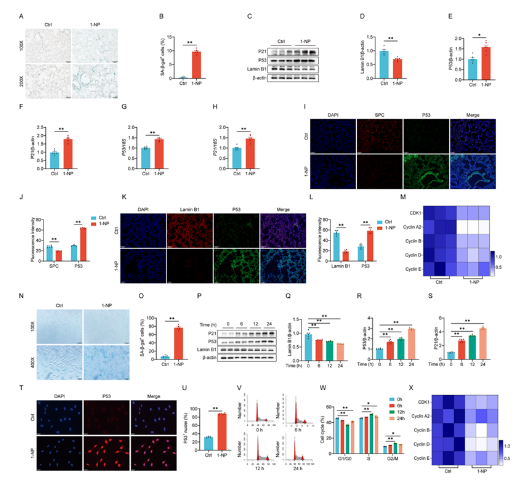
2. 1-NP暴露誘導小鼠肺和MLE-12細胞周期停滯與早衰
1-NP暴露使小鼠肺組織中SA-β-gal陽性細胞增多,Lamin B1表達降低,p53、p21的mRNA和蛋白水平升高,肺泡Ⅱ型細胞中p53與SPC共定位增加;MLE-12細胞中同樣出現SA-β-gal陽性細胞增多、細胞周期S/G2/M期阻滯、p53/p21通路激活及SASP因子表達上調,表明1-NP誘導肺上皮細胞衰老。
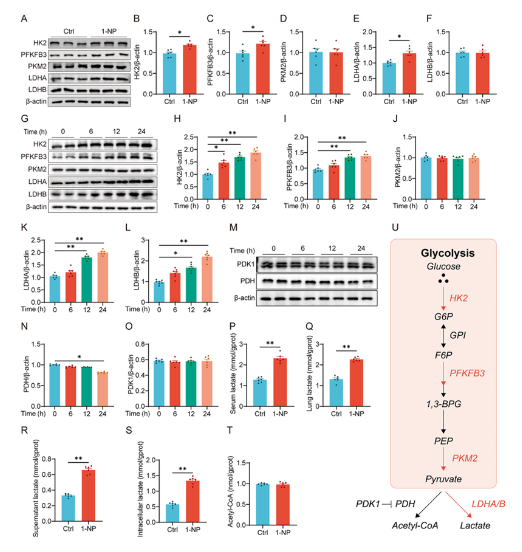
3. 1-NP暴露促進小鼠肺和MLE-12細胞糖酵解與乳酸生成
1-NP暴露上調小鼠肺和MLE-12細胞中糖酵解關鍵酶(HK2、PFKFB3、LDHA)的表達,促進乳酸生成,而丙酮酸脫氫酶(PDH)表達降低,乙酰fu酶A水平無顯著變化,提示1-NP通過增強糖酵解和LDHA活性導致乳酸積累。
4. 1-NP暴露誘導肺上皮細胞組蛋白乳酸化而非乙酰化
1-NP暴露顯著增加小鼠肺組織和MLE-12細胞中Pan Kla、H3K14la等組蛋白乳酸化水平,且呈時間和劑量依賴性,但組蛋白乙酰化(如H3K14ac)水平無明顯變化,表明1-NP特異性誘導組蛋白乳酸化修飾。

5. LDHA敲低減輕1-NP誘導的組蛋白乳酸化、細胞周期停滯和早衰
通過siRNA敲低LDHA可減少1-NP誘導的乳酸生成,降低H3K14la水平,緩解MLE-12細胞的細胞周期阻滯、SA-β-gal陽性細胞增多及p53/p21通路激活,抑制SASP因子分泌,證實LDHA介導的乳酸生成是組蛋白乳酸化和細胞衰老的關鍵上游事件。

6. 組蛋白乳酸化激活肺上皮細胞p53轉錄
CUT&Tag和ChIP-qPCR實驗顯示,1-NP誘導的H3K14la在p53啟動子區富集,直接促進p53轉錄;敲低p53可逆轉1-NP引起的p21表達上調、細胞周期停滯和衰老,提示H3K14la通過激活p53轉錄驅動細胞衰老。

7. OXA補充緩解1-NP誘導的小鼠肺細胞周期停滯和衰老
LDH抑制劑oxamate(OXA)預處理可降低1-NP暴露小鼠的血清和肺組織乳酸水平,抑制H3K14la表達,減少SA-β-gal陽性細胞和p53/p21通路激活,緩解細胞周期停滯和SASP因子釋放,表明抑制乳酸生成可減輕1-NP誘導的肺上皮細胞衰老。
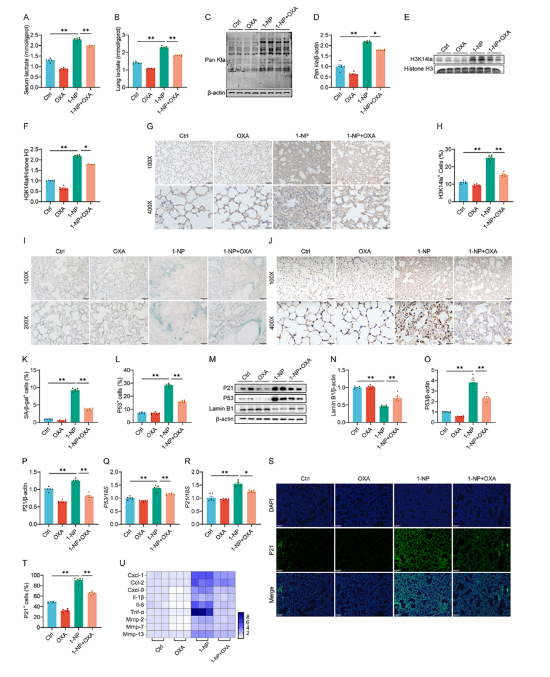
8. OXA補充減輕1-NP誘導的小鼠COPD樣表型
OXA預處理顯著改善1-NP暴露小鼠的肺重量、肺系數及肺泡結構損傷,降低氣道壁厚度和炎癥細胞浸潤,改善肺功能指標(FVC、FEV1、FEV1/FVC、PEF),提示抑制乳酸生成可緩解1-NP誘導的COPD樣病理改變。
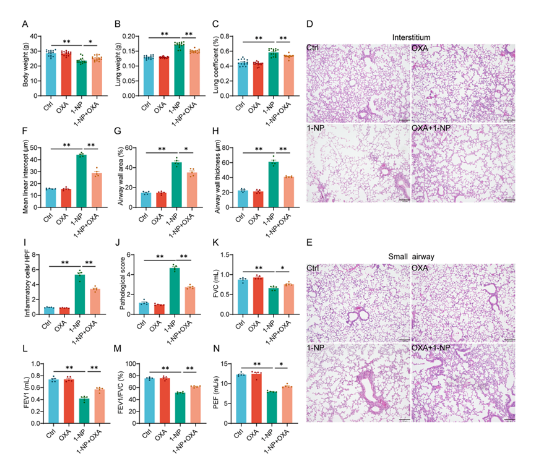
研究結論:
慢性1-NP暴露通過上調肺上皮細胞糖酵解和LDHA表達,增加乳酸生成,誘導H3K14la,進而激活p53轉錄,導致細胞周期停滯和早衰,最終引發COPD。抑制乳酸生成可減輕1-NP介導的上述病理過程,提示糖酵解-乳酸-H3K14la-p53軸是COPD潛在治療靶點。
研究的創新性:
揭示組蛋白乳酸化(H3K14la)在1-NP誘導COPD中的作用,證實H3K14la通過直接激活p53轉錄調控肺上皮細胞衰老,為環境污染物致COPD的表觀遺傳機制提供新見解。
研究的不足之處:
僅聚焦肺上皮細胞,未探討1-NP對其他肺細胞(如成纖維細胞、巨噬細胞)的影響;未明確1-NP誘導糖酵解上調的上游信號;動物模型暴露方式與人類實際暴露場景可能存在差異。
研究展望:
后續可探究1-NP對肺組織其他細胞類型的作用及機制;挖掘調控糖酵解的上游分子(如HIF-1α)在1-NP致COPD中的作用;開展人群流行病學研究,驗證H3K14la和p53在環境污染物暴露相關COPD患者中的臨床意義;開發靶向LDHA或H3K14la的特異性抑制劑用于COPD治療。
研究意義:
揭示環境污染物1-NP致COPD的新機制,拓展對COPD發病的認知;提出組蛋白乳酸化和乳酸生成作為COPD治療新靶點,為開發抗環境污染物相關COPD的藥物提供理論依據;強調控制1-NP等環境污染物暴露對COPD預防的重要性。



